

Translate this page into:
A rare case of Type 4 Pachyonychia congenita
*Corresponding author: Dr. Suhail Khan MK, Department of Dermatology, Bangalore Medical College and Research Institute, Bengaluru, Karnataka, India. suhailkhan71125@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Khan MS, Mohan HN, Shilpa K, Eswari L. A rare case of Type 4 Pachyonychia congenita. J Onychol Nail Surg. doi: 10.25259/JONS_22_2024
Abstract
Pachyonychia congenita (PC) is an uncommon keratinisation disorder with autosomal dominant inheritance. This inherited ectodermal abnormality is characterised by pathognomonic nail deformities and defective keratin production. It typically manifests at birth or develops within a year of birth, as the name suggests. The clinical diagnostic hallmark of PC is the presence of thickened wedge-shaped nails. PC tarda (PCT) is a distinct variant of PC characterised by later onset, usually in the second and third decade of life. Very few cases have been reported in the literature. According to the recent classification of PC, our case had the features of Type 4-PC-K16 (caused by pathogenic variation in KRT16). This case is being reported for its rarity as the patient had onset of lesions at 18 years of age, and to corroborate findings of previous report where sparing of lunula was proposed as a clinical sign to differentiate PCT from other causes of subungual hyperkeratosis.
Keywords
Pachyonychia congenita tarda
Thickened nails
Follicular papules
Lunula
Type 4 Pachyonychia congenita

INTRODUCTION
The term Pachyonychia congenita (PC) was first coined by Jadassohn and Lewandowsky in 1906. It is used to describe a class of uncommon ectodermal dysplasias that are typically inherited in an autosomal dominant fashion. Nonetheless, reports of sporadic and autosomal recessive cases are found in the literature.[1,2]
PC is associated with mutations in keratin genes K6a, K6b, K16 or K17.[3] It is characterised by an early onset of nail dystrophy with a defect in the keratinisation of mucosa and skin.[4]
Most of the reports documented in literature have had an early onset during infancy, irrespective of the disorder’s sub-classification or mode of inheritance. According to the old classification, late-onset PC was regarded as rare with very few cases having been reported. The term PC tarda (PCT) has been suggested to describe this late-onset form of PC.[4] Similar underlying genetic abnormalities affecting the keratin 6a/16 pair may be present in PCT, which is believed to be a delayed version of PC type 1.[5]
According to the recent classification, PC is classified into five types based on the mutated gene. PC-K16 (caused by pathogenic mutations in KRT16) is Type 4 PC and characterised mainly by fingernail and toenail thickening with late onset (>15 years of age).[6]
CASE REPORT
A 20-year-old male, born to parents with 3rd-degree consanguinity, presented to the dermatology outpatient department with complaints of nail thickening and elevated skin lesions all over the body for the past 2 years. Nail thickening initially started over few of the fingernails and gradually progressed to involve all 20 nails over a period of 2 years. The patient also noticed asymptomatic raised skin-coloured lesions 6 months following the onset of nail lesions. They initially started over both upper limbs and gradually progressed to involve the trunk, back, buttocks and both lower limbs. There was no history of palmoplantar lesions, palmoplantar pain, hyperhidrosis, oral lesions, dental abnormalities and hoarseness of voice or ocular complaints. On examination, all 20 nails showed discoloration and thickening with massive subungual hyperkeratosis, producing a distal elevation of the nail plate and wedge-shaped deformity [Figure 1]. Keratotic follicular papules were noted diffusely over the back, trunk and bilateral upper and lower limb [Figure 2]. Callosities were noted over bilateral malleoli. Palms and soles were normal. Oral cavity examination was normal. Onychoscopic examination revealed compact subungual hyperkeratosis with nail plate thickening [Figure 3].
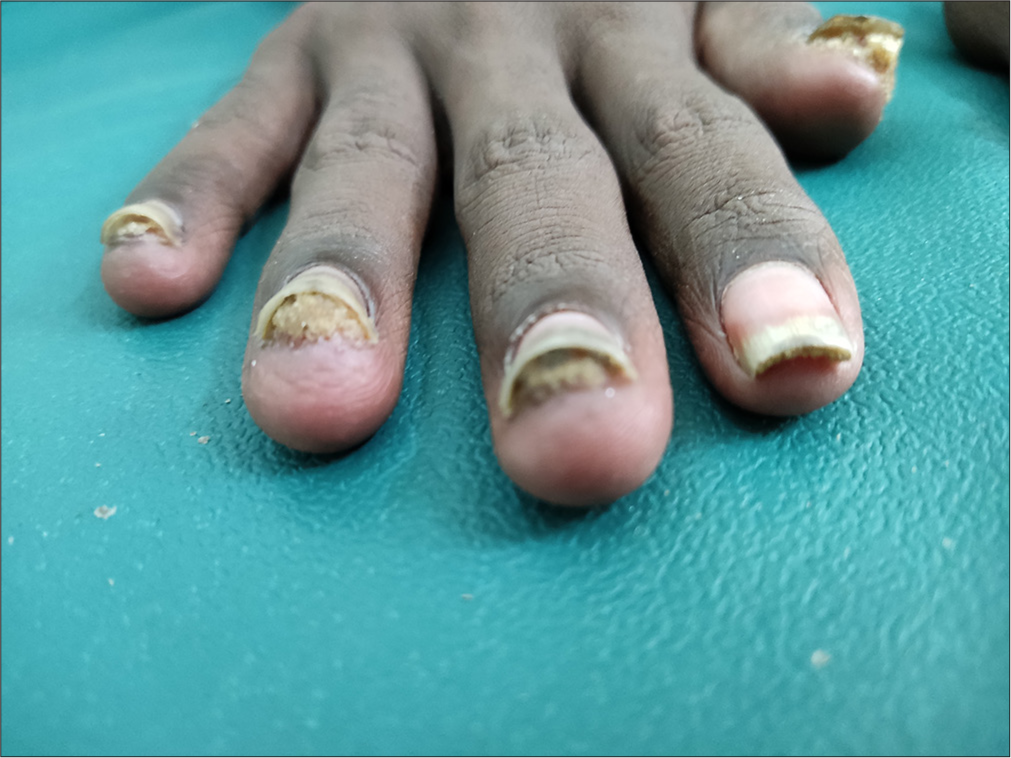
- Discoloured and thickened fingernails with massive subungual hyperkeratosis producing a distal elevation of nail plate and wedge-shaped deformity of the nails.
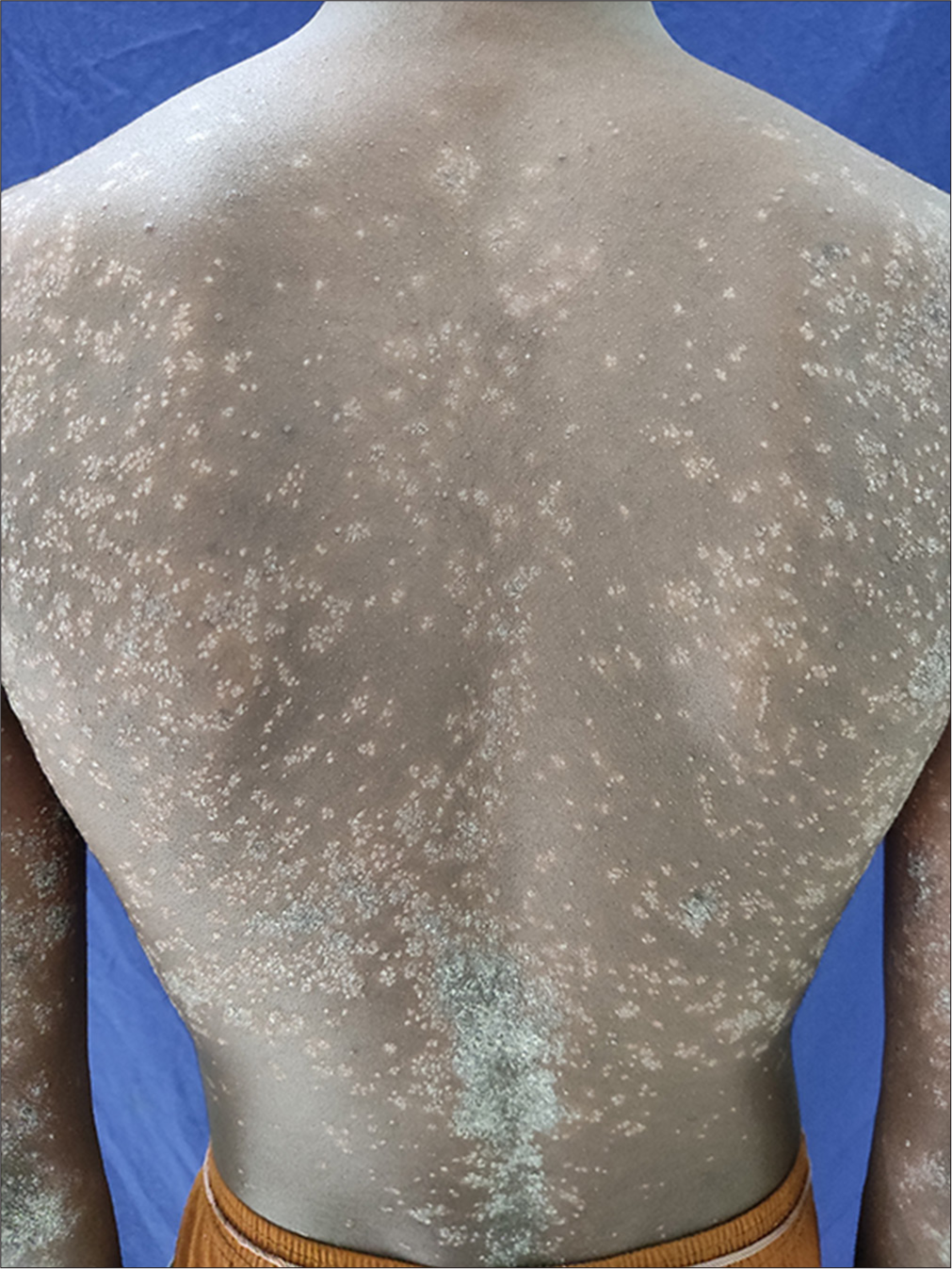
- Multiple hyperkeratotic follicular papules involving trunk.
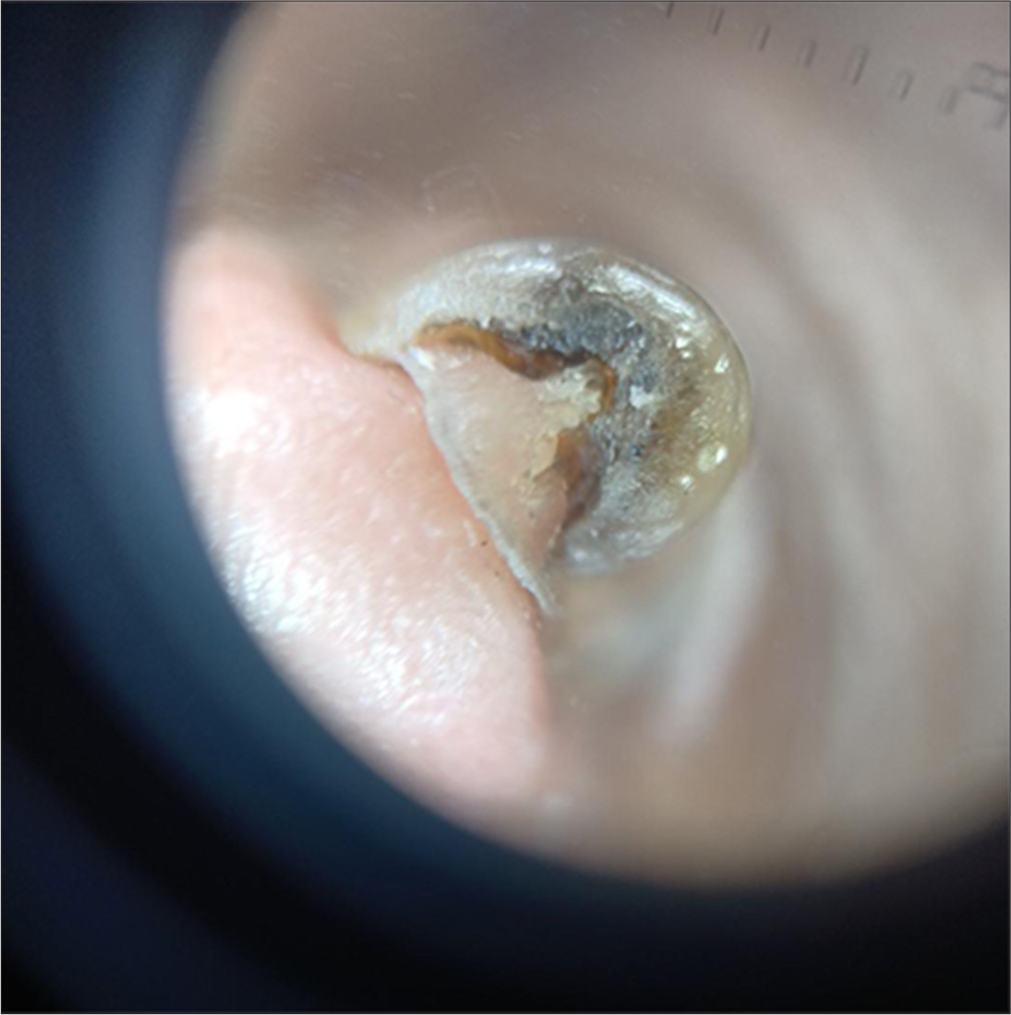
- Onychoscopy image showing compact subungual hyperkeratosis with thickened and discoloured nail plate with wedge-shaped deformity.
Potassium hydroxide mount from nail and mucosal scrapings was negative for fungal disease. All routine investigations were within normal limits. Histopathological examination from the keratotic follicular papule revealed orthokeratotic hyperkeratosis and acanthosis [Figure 4]. The patient did not consent for a nail biopsy.
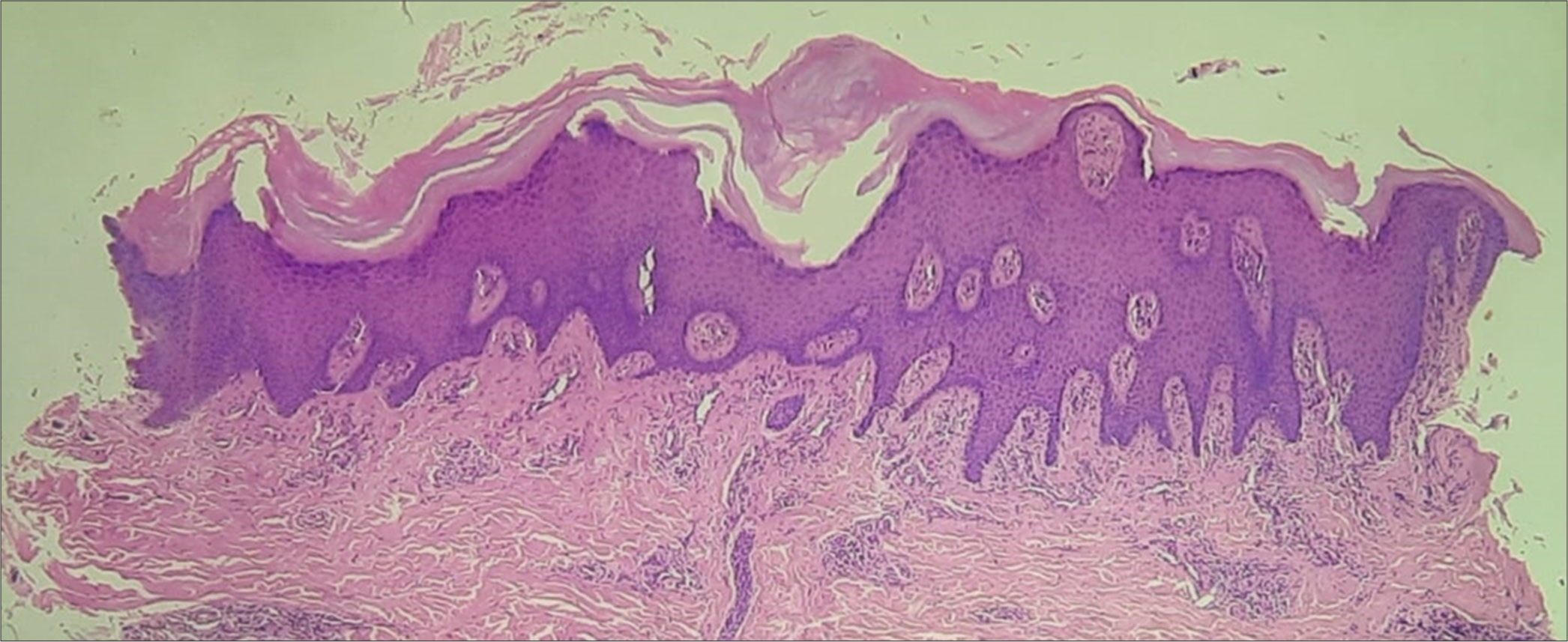
- Skin biopsy from keratotic papule showing orthokeratotic hyperkeratosis, acanthosis with mild superficial inflammatory infiltrate. (H&E stain, 20× magnification).
Since our case had both fingernail and toenail thickening along with late-onset, a diagnosis of PC-K16 (caused by pathogenic variants in KRT16) was made.
DISCUSSION
Wilson originally described PC in 1905 and JadassohnLewandowsky described the association between PC and ectodermal abnormalities.[1,7,8] The Schonfeld classification is the most common classifications.[9] With detailed clinical history and pathogenic variants being identified in an increasing number of people with PC, it was evident that the older classification was not applicable to the broader spectrum of PC. Recent classification divides PC into five types based on the keratin gemer mutation.
Type 1-PC-K6a (mutations in KRT6A gene)
Type 2-PC-K6b (mutations in KRT6B gene)
Type 3-PC-K6c (mutations in KRT6C gene)
Type 4-PC-K16 (mutations in KRT16 gene)
Type 5-PC-K17 (mutations in KRT17 gene).[6]
Our case was classified as Type 4 PC as he had both finger and toe nail thickening and the onset was at 18 years of age. Mutation analysis could not be done due to the financial constraints.
The characteristic nail changes of transverse over-curvature, distal onycholysis, subungual hyperkeratosis and variable discoloration are common to all types of PC. Type 1 (Jadassohn–Lewandowsky, PC-1) also consists of hyperkeratosis of palms, soles, knees and elbows, follicular hyperkeratosis and oral leukokeratosis. Rarely, blister formation, hoarse voice due to laryngeal involvement and hyperhidrosis can be seen. In type 2 (Jackson–Lawler, PC-2), the palmoplantar keratoderma (PPK) and oral changes are less marked or absent. In addition, a history of natal teeth and the development of epidermal cysts or steatocysts are found. Type 3 (Schafer–Brunauer, PC-3) has similar features to PC-1 with additional leukokeratosis of the cornea. A fourth form in which the typical nail changes begins in the second and third decades of life, had been described as PCT.[10]
Treatment options include keratolytics and emollients for hyperkeratotic skin and nail changes. Oral retinoids have been reported to aggravate the PPK, subungual hyperkeratosis and the keratotic patches on the knees and in the mouth. Surgical modalities of treatment include nail removal and various methods to ablate the nail matrix.[11]
Other differentials considered were onychomycosis, Twenty-nail dystrophy, dyskeratosis congenita (DC), Olmsted syndrome, psoriasis and lichen planus. Dermatophytic infections do not affect all fingers and toes simultaneously, especially in young children. Twenty-nail dystrophy occurs without keratoderma or other associated changes. Autosomal dominant inheritance has been described. Features of DC, such as nail dystrophy, PPK, hyperhidrosis and oral leukoplakia, can mimic those of PC. However, haematologic manifestations skin tumours and reticulate hyperpigmentation are distinguishing characteristics that were absent in our case. Painful PPK is a hallmark of Olmsted syndrome. Other findings include periorificial keratotic plaques and constricted digital bands, leading to spontaneous amputation, mutilating PPK, nail dystrophy, alopecia and itching. Psoriasis and Lichen planus do not involve all 20 nails and are characteristic of clinical and histopathological features which were absent in our case.
According to Bahhady et al., sparing of the lunula is one of the important clinical clues to differentiate PCT from other causes of subungual hyperkeratosis. It is seen as the keratin 6a and 16 are expressed in the nail bed producing changes in the form of subungual hyperkeratosis and nail plate dystrophy.[2] Our case also demonstrated similar changes of subungual hyperkeratosis with the sparing of lunula in most of the nails [Figure 4].
CONCLUSION
Type 4-PC should be kept in mind when facing severe subungual hyperkeratosis affecting all 20 nails, regardless of the age of the patient. The recent classification of PC should be employed based on the keratin gene mutation. Sparing of the lunula can help differentiate Type 4-PC from other causes of subungual hyperkeratosis.
Author’s contributions
Each author helped with the study’s design and manuscript preparation, data collection, analysis, and interpretation, article drafting, clinical revisions and final approval of the submitted version.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Pachyonychia congenita: Iknografia Dermatological Berlin: Urban and Schwarzenberg; 1906. p. :29-31.
- [Google Scholar]
- Pachyonychia congenita tarda: Very late onset. Int J Dermatol. 2008;47:1172-3.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and pathological features of pachyonychia congenita. J Investig Dermatol Symp Proc. 2005;10:3-17.
- [CrossRef] [PubMed] [Google Scholar]
- Pachyonychia congenita tarda: A rare case report. Contemp Clin Dent. 2013;4:409-11.
- [CrossRef] [PubMed] [Google Scholar]
- Subungual hyperkeratosis. In: The nail indifferential diagnosis. United Kingdom: Informa Healthcare; 2006. p. :51-60.
- [CrossRef] [Google Scholar]
- Pachyonychia congenita In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993-2024.
- [Google Scholar]
- Three cases of hereditary hyperkeratosis of the nail-bed. Arch Dermatol Syphilol. 1905;2:13-4.
- [Google Scholar]
- The pachyonychia congenita syndrome. Acta Derm Venerol. 1980;60:45-9.
- [CrossRef] [PubMed] [Google Scholar]
- Pachyonychia congenita tarda. A late-onset form of pachyonychia congenita. Arch Dermatol. 1991;127:701-3.
- [CrossRef] [PubMed] [Google Scholar]
- Pachyonychia congenita tarda. Australas J Dermatol. 2000;41:175-7.
- [CrossRef] [PubMed] [Google Scholar]




